
Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
Sindrome di Treacher Collins
Esperto medico dell'articolo

Ultima recensione: 04.07.2025

I disturbi intrauterini nei processi di sviluppo osseo causano gravi deformità craniofacciali e una delle varianti di tale patologia è la sindrome di Treacher Collins (TCS) o disostosi mandibolo-fasciale, cioè maxillo-facciale.
Codice della malattia secondo ICD 10: classe XVII (anomalie congenite, deformazioni e disturbi cromosomici), Q75.4 - disostosi mandibolofacciale.
Le cause Sindrome di Treacher Collins
Questa sindrome prende il nome dall'eminente oculista britannico Edward Treacher Collins, che ne descrisse le caratteristiche principali più di cento anni fa. Tuttavia, i medici europei chiamano più spesso questo tipo di anomalia delle ossa facciali e mascellari "malattia o sindrome di Franceschetti", sulla base delle approfondite ricerche dell'oculista svizzero Adolf Franceschetti, che coniò il termine "disostosi mandibolo-fasciale" a metà del secolo scorso. In ambito medico, viene utilizzato anche il nome di sindrome di Franceschetti-Collins.
La sindrome di Treacher Collins è causata da mutazioni nel gene TCOF1 (localizzato sul cromosoma 5q31.3-33.3), che codifica per una fosfoproteina nucleolare responsabile della formazione della porzione craniofacciale dell'embrione umano. A causa di una riduzione prematura della quantità di questa proteina, la biogenesi e le funzioni dell'rRNA vengono interrotte. Secondo i genetisti del programma di ricerca sul Genoma Umano, questi processi portano a una riduzione della proliferazione delle cellule embrionali della cresta neurale, una cresta lungo il solco neurale che si chiude in un tubo neurale durante lo sviluppo embrionale.
La formazione dei tessuti facciali avviene grazie alla trasformazione e alla differenziazione delle cellule della parte superiore (testa) della cresta neurale, che migrano lungo il tubo neurale fino all'area del primo e del secondo arco branchiale dell'embrione. La carenza di queste cellule causa deformazioni craniofacciali. Il periodo critico per la comparsa di anomalie è compreso tra 18 e 28 giorni dopo la fecondazione. Al termine della migrazione delle cellule della cresta neurale (nella quarta settimana di gestazione), si formano quasi tutti i tessuti mesenchimali lassi nell'area facciale, che successivamente (tra le 5 e le 8 settimane) si differenziano nei tessuti scheletrici e connettivi di tutte le parti del viso, del collo, della laringe, dell'orecchio (incluso l'orecchio interno) e dei futuri denti.
Patogenesi
La patogenesi della sindrome di Treacher Collins è spesso familiare e l'anomalia è ereditata con modalità autosomica dominante, sebbene esistano casi di trasmissione autosomica recessiva del difetto (con mutazioni in altri geni, in particolare POLR1C e POLR1D). L'aspetto più imprevedibile della disostosi maxillo-facciale è che la mutazione viene ereditata dai figli solo nel 40-48% dei casi. Ciò significa che nel 52-60% dei pazienti le cause della sindrome di Treacher Collins non sono associate alla presenza di un'anomalia in famiglia e si ritiene che la patologia si verifichi a seguito di mutazioni genetiche sporadiche de novo. Molto probabilmente, le nuove mutazioni sono la conseguenza di effetti teratogeni sul feto durante la gravidanza.
Tra le cause teratogene di questa sindrome, gli esperti citano dosi elevate di etanolo (alcol etilico), radiazioni, fumo di sigaretta, citomegavirus e toxoplasma, nonché erbicidi a base di glifosato (Roundal, Glyfor, Tornado, ecc.). L'elenco dei fattori iatrogeni include farmaci per l'acne e la seborrea a base di acido 13-cis-retinoico (Isotretinoina, Accutane); il farmaco anticonvulsivante Fenitoina (Dilantin, Epanutin); psicofarmaci Diazepam, Valium, Relanium, Seduxen.
Sintomi Sindrome di Treacher Collins
Nella maggior parte dei casi, i segni clinici della disostosi mandibolo-fasciale e il grado della loro espressione dipendono dalle caratteristiche della manifestazione delle mutazioni genetiche. E i primi segni di questa anomalia, nella maggior parte dei casi, sono visibili in un bambino subito dopo la nascita: il viso con sindrome di Treacher Collins presenta un aspetto caratteristico. Inoltre, le anomalie morfologiche sono solitamente bilaterali e simmetriche.
I sintomi più evidenti della sindrome di Treacher Collins sono:
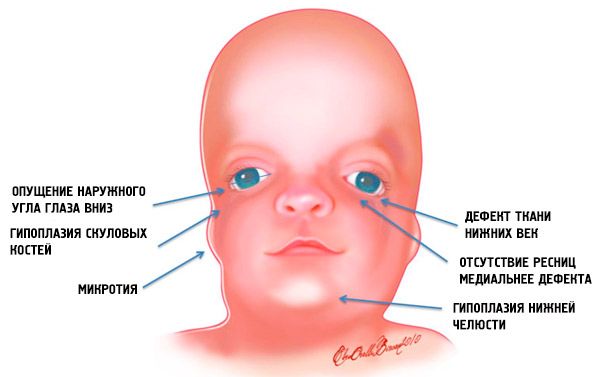
- sottosviluppo (ipoplasia) delle ossa facciali del cranio: zigomatico, processi zigomatici dell'osso frontale, lamine pterigoidee laterali, seni paranasali, mandibola e protrusioni delle epifisi ossee (condili);
- sottosviluppo delle ossa della mandibola (micrognazia) e angolo mandibolare più ottuso del normale;
- il naso è di dimensioni normali, ma appare grande a causa dell'ipoplasia delle arcate sopraccigliari e del sottosviluppo o dell'assenza delle arcate zigomatiche nella regione temporale;
- le fessure oculari sono rivolte verso il basso, cioè la forma degli occhi è anormale, con gli angoli esterni che pendono verso il basso;
- difetti delle palpebre inferiori (coloboma) e assenza parziale delle ciglia su di esse;
- padiglioni auricolari di forma irregolare con un'ampia gamma di deviazioni, tra cui la loro posizione nell'angolo della mascella inferiore, l'assenza di lobi, fistole cieche tra il trago dell'orecchio e l'angolo della bocca, ecc.;
- restringimento o chiusura (atresia) del condotto uditivo esterno e anomalie degli ossicini dell'orecchio medio;
- assenza o ipoplasia delle ghiandole salivari parotidi;
- ipoplasia faringea (restringimento della faringe e delle vie aeree);
- mancata fusione del palato duro (palatoschisi), nonché assenza, accorciamento o immobilità del palato molle.
Tali anomalie anatomiche presentano in ogni caso delle complicazioni. Si tratta di disturbi uditivi funzionali, che si manifestano con ipoacusia trasmissiva o sordità completa; deficit visivi dovuti a una formazione anomala dei bulbi oculari; difetti del palato che causano difficoltà di alimentazione e deglutizione. Esistono disturbi dell'occlusione dentale (malocclusione) associati a difetti mandibolari, che a loro volta causano problemi di masticazione e articolazione. Le patologie del palato molle spiegano la voce nasale.
Complicazioni e conseguenze
Le conseguenze delle anomalie maxillo-facciali nella sindrome di Treacher Collins sono che alla nascita le capacità intellettive del bambino sono normali, ma a causa di difetti dell'udito e altri disturbi, si osserva un ritardo mentale secondario.
Inoltre, i bambini con tali difetti avvertono acutamente la loro inferiorità e soffrono, il che influisce negativamente sul loro sistema nervoso e sulla loro psiche.
Diagnostica Sindrome di Treacher Collins
La diagnosi postnatale della sindrome di Treacher Collins si basa essenzialmente sui segni clinici. La disostosi craniofacciale è facilmente identificabile quando la sindrome è pienamente manifesta, ma quando sono presenti sintomi patologici minimamente espressi, possono sorgere difficoltà nel formulare una diagnosi corretta.
In questo caso, è necessario prestare particolare attenzione alla valutazione di tutte le funzioni associate alle anomalie, in particolare quelle che interessano la respirazione (a causa del rischio di apnea notturna). È inoltre necessario valutare e monitorare l'efficacia dell'alimentazione e la saturazione dell'ossigeno dell'emoglobina.
Successivamente, tra il 5° e il 6° giorno di vita, sarà necessario accertare l'entità del danno uditivo mediante un esame audiologico, da effettuare presso la maternità.
Viene prescritta una visita durante la quale si esegue una diagnostica strumentale mediante fluoroscopia della dismorfologia craniofacciale; pantomografia (radiografia panoramica delle strutture ossee del cranio facciale); tomografia computerizzata cranica completa in varie proiezioni; TC o RM dell'encefalo per determinare lo stato del condotto uditivo interno.
La diagnosi più precoce (prenatale) di anomalie maxillo-facciali in presenza di sindrome di Treacher Collins nella storia familiare è possibile mediante biopsia dei villi coriali alla 10a-11a settimana di gravidanza (la procedura comporta il rischio di aborto spontaneo e di infezione dell'utero).
Vengono inoltre effettuati esami del sangue ai familiari; alla 16a-17a settimana di gravidanza si analizza il liquido amniotico (amniocentesi transaddominale); alla 18a-20a settimana di gravidanza si esegue una fetoscopia, ovvero un prelievo di sangue dai vasi fetali della placenta.
Ma il più delle volte, per la diagnosi prenatale di questa sindrome nel feto (tra la 20a e la 24a settimana di gravidanza) si ricorre all'ecografia.
Quali test sono necessari?
Diagnosi differenziale
Questi stessi metodi vengono utilizzati dagli specialisti quando è necessaria una diagnosi differenziale per riconoscere la sindrome di Treacher Collins lieve e distinguerla da altre anomalie congenite delle ossa craniofacciali, in particolare: sindromi di Apert, Crouzon, Nager, Peters-Hewels, Hellermann-Steph, nonché microsomia emifacciale (sindrome di Goldenhar), ipertelorismo, fusione prematura delle suture craniche (craniosinostosi) o fusione alterata delle ossa facciali (craniosinostosi).
Trattamento Sindrome di Treacher Collins
Come in tutti i casi di difetti congeniti geneticamente determinati, il trattamento delle forme gravi della sindrome di Treacher Collins è esclusivamente palliativo, poiché non esistono metodi terapeutici per tali patologie. Lo spettro e il grado di malformazioni in questa sindrome sono ampi e, pertanto, anche la natura e l'intensità dell'intervento medico offrono numerose opzioni.
Per correggere e migliorare l'udito si utilizzano apparecchi acustici, mentre per migliorare la parola si ricorre a sedute di logopedia.
Interventi chirurgici sono necessari in età precoce nei casi gravi di restringimento delle vie aeree (si esegue una tracheostomia) e della laringe (si esegue una gastrostomia per l'alimentazione). Può essere necessaria anche la correzione chirurgica del palato.
Gli interventi di allungamento mandibolare vengono eseguiti a partire dai 2-3 anni di età o più tardi. La ricostruzione dei tessuti molli include la correzione del coloboma della palpebra inferiore e la chirurgia plastica auricolare.
Prevenzione
Previsione
Qual è la prognosi di questa patologia? Dipende dal grado di deformazione e dall'intensità dei sintomi. La sindrome di Treacher Collins è una diagnosi che dura tutta la vita.
 [ 25 ]
[ 25 ]